
What is gene therapy, and is your hospital prepared to handle gene therapy products?
ASHP Advantage is coordinating an educational initiative aimed at answering those questions. On the initiative website you’ll find both live and on-demand continuing education activities to help you become knowledgeable regarding the clinical uses, operational considerations, and safe handling of gene therapies.
This e-newsletter provides an overview of the different types of gene therapy, challenges and concerns related to gene therapies, and the role of the Food and Drug Administration (FDA) and guidelines in product development. The second e-newsletter in this series will highlight several new developments in gene therapy and address operational and safe-handling considerations.
The educational initiative draws on the expertise of two pharmacists who are involved with this cutting-edge area of practice. John Petrich, B.S.Pharm., M.S., is Manager of the Investigational Drug Service at Cleveland Clinic in Ohio. He first became involved with gene therapy approximately 12 years ago when clinical trials of two gene therapy products involving different vectors raised new issues related to product handling, standard operating procedures, and staff training. Mr. Petrich was instrumental in addressing these concerns, and his work on subsequent clinical trials laid the groundwork for future clinical trials and integration of gene therapy products into the health system.
For Michael Storey, Pharm.D., M.S., BCPS, the path to becoming involved with gene therapy began later and involved commercial gene therapy products. Although he joined the pharmacy staff at Nationwide Children’s Hospital in Columbus, Ohio, as a manager of the oncology pharmacy and other clinical pharmacy services in 2012, he did not become involved in gene therapy until several years ago when he assumed the role of Medication Use and Formulary Coordinator. About that time, FDA approved nusinersen for spinal muscular atrophy, and he worked with the neuromuscular team in rolling out that product throughout the organization. He has also collaborated with his institution’s gene and cell therapy teams to prepare the institution for commercial gene replacement and cellular therapy.
“That model of developing relationships with the teams who provide care to patients who benefit from the different gene therapies, as well as with hospital finance, has been instrumental in preparing for commercialization of these products,” says Dr. Storey.


What is Gene Therapy?
Many diseases and medical conditions are attributed to the absence or dysfunction of a gene that encodes production of a vital protein (Figure).
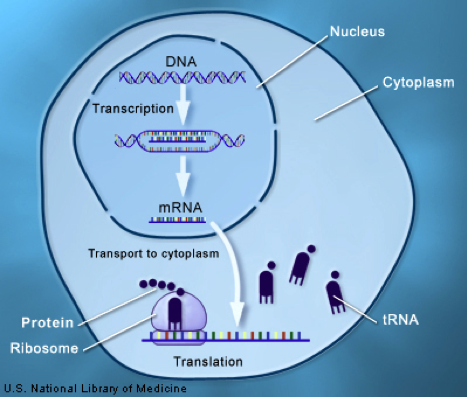
Gene therapy is defined as a therapy that acts on or modifies a gene for therapeutic benefit. Gene therapies can provide benefit through several mechanisms:
- Ex vivo genetic modification of cells for a therapeutic purpose (cellular gene therapy),
- Modifying activity of a gene that does not function properly (regulators of gene expression and gene regulation),
- Introducing a new or modified gene to treat a disease (gene replacement therapy), or
- Direct editing of the genome to remove defective genes, insert functional genes, or both.
Gene therapy differs from genomic medicine, which entails the use of genomic information about a patient as part of the diagnostic or therapeutic decision-making process.
The following table shows examples of gene therapy products approved by FDA for different types of gene therapy.
| (Therapeutic Class / FDA Approval) |
|
|---|---|
| Cellular Gene Therapies | |
| Tisagenlecleucel (Oncology / 2017) | |
| Axicabtagene ciloleucel (Oncology /2017) | |
| Regulators of gene expression: antisense oligonucleotides | |
| Mipomersen (Dyslipidemia / 2013) | |
| Eteplirsen (Neuromuscular /2016) | |
| Nusinersen (Neuromuscular /2016) | |
| Regulators of gene expression: double-stranded RNA | |
| Inotersen (Neurology / 2018) | |
| Patisiran (Neurology /2018) | |
| Gene replacement therapy | |
| Voretigene neparvovec-rzyl (Ophthalmology / 2017) | |
| Products | Therapeutic Class | FDA Approval |
|---|---|---|
| Cellular gene therapies | ||
| Tisagenlecleucel | Oncology | 2017 |
| Axicabtagene ciloleucel | Oncology | 2017 |
| Regulators of gene expression: antisense oligonucleotides | ||
| Mipomersen | Dyslipidemia | 2013 |
| Eteplirsen | Neuromuscular | 2016 |
| Nusinersen | Neuromuscular | 2016 |
| Regulators of gene expression: double-stranded RNA | ||
| Inotersen | Neurology | 2018 |
| Patisiran | Neurology | 2018 |
| Gene replacement therapy | ||
| Voretigene neparvovec-rzyl | Ophthalmology | 2017 |
The anti-CD19 chimeric antigen receptor (CAR)-T cell therapy products tisagenlecleucel and axicabtagene ciloleucel are approved by FDA for certain relapsed or refractory leukemias and lymphomas. These cellular gene therapies are produced ex vivo by performing leukapheresis to harvest a patient’s T-cells and using a viral vector to genetically modify the T-cells so that they engage antigens expressed on tumor cells after reinfusion of the modified T-cells into the patient.
Regulators of gene expression include antisense oligonucleotides (e.g., mipomersen, eteplirsen, nusinersen) and double-stranded RNA (e.g., inotersen, patisiran). These therapies exert their therapeutic effects by altering DNA transcription, which affects protein synthesis. They are small-molecule, nonhazardous drugs.
Gene replacement therapies involve the use of a viral capsid (i.e., vector) to deliver genetic material (a transgene) to the cellular nucleus for the treatment of monogenic diseases, which are diseases caused by mutation or deletion of a single gene. The empty viral capsid elicits and is eliminated by an immune response. Voretigene neparvovec-rzyl was the first gene replacement therapy approved by FDA in December 2017 for the treatment of an inherited retinal disease that causes blindness.
Systemic gene replacement therapies have been evaluated in clinical trials but they are not yet approved by FDA. Agents that cross the blood-brain barrier and gain access to the central nervous system may be advantageous for treating certain diseases, such as spinal muscular atrophy. As one might anticipate after administering a viral vector, systemic adverse effects (e.g., transient transaminitis, arthralgia, headache, myalgia, back pain, fatigue, insomnia, infections) may develop. Prophylactic and symptomatic treatment may be warranted for some of these effects.
Clustered Regularly Interspaced Short Palindromic Repeats-Associated Protein 9 (known as CRISPR-Cas9) gene therapies permit gene editing at specific sites in the genome. This technology facilitates modification of large DNA segments to produce a therapeutic effect. None of these products are approved by FDA at this time, and approval is not likely within the next 5 years because these products are in the preclinical or early clinical phase of development.
More Information
- U.S. Food and Drug Administration. What is gene therapy? July 25, 2018. (accessed 2019 Feb 1).
- Wang D, Gao G. State-of-the-art human gene therapy: part I. Gene delivery technologies. Discov Med. 2014; 18:67-77.
- Mendell JR, Al-Zaidy S, Shell R et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017; 377:1713-22.
Challenges and Concerns
Balancing efficacy, safety, and cost is a challenge in developing and using gene therapy products. Concerns associated with the use of gene therapies include the adequacy and durability of response and unintended effects (i.e., unwanted effects mediated elsewhere in the genome or in cells other than those targeted). The durability of response is particularly important for patients who are very young when they receive gene therapy. Transgene expression can be unstable and limit the durability of response to gene therapy, so gene promoters have been used to maintain the function of transgenes. Although the early results of studies of the durability of response to gene therapy are promising, additional data from research for longer follow-up periods are needed to verify that the clinical response is sustained.
Seroconversion is another concern associated with the use of gene therapies. Seroconversion is the formation of neutralizing antibodies in response to exposure to a viral vector used in a gene therapy product. Some patients are seropositive to the viruses used for gene therapy products before exposure to those products, and others may become seropositive as a result of exposure. Seropositive patients were excluded from many clinical trials of gene therapy products.
Seroconversion in patients during treatment would be expected to preclude repeat use of gene therapy with the same viral vector because of the limited availability of interventions to overcome seropositivity. Seroconversion can occur in healthcare providers as a result of occupational exposure during the administration of gene therapy products or patient care tasks performed after administration (i.e., from exposure to bodily fluids). A patient’s family members and caregivers also could experience seroconversion. Seroconversion in healthcare providers, family members, and caregivers could be problematic if it precludes the use of gene therapy in these individuals.
Cost is a consideration because gene therapies are expensive to manufacture and often useful for only very small numbers of patients. Illnesses treated with gene therapy are often also costly to manage without gene therapy. The potential for one-time treatment with some gene therapy products instead of multiple doses over a long period to treat a chronic disease should be taken into consideration when performing economic analyses. Early and frequent engagement with stakeholders is among the strategies used by health system pharmacists to manage the cost of gene therapies.
More Information
- Deverman BE, Ravina BM, Bankiewicz KS et al. Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov. 2018; 17:641-59.
Role of FDA and Guidelines for Product Development
Gene therapy products are biological products regulated by the FDA Center for Biologics Evaluation and Research. Submission of an investigational new drug application to FDA is required before performing clinical studies in humans. Submission and approval of a biologics license application is required for marketing of gene therapy products in the United States.
In July 2018, FDA issued draft guidance with recommendations for the design of long-term follow-up observational studies to collect data on delayed adverse events following the administration of gene therapy products. Because gene therapies produce therapeutic effects through permanent or long-acting changes to the body, unanticipated adverse events could develop long after gene therapy administration. The draft guidance addresses product characteristics, patient-related factors, and preclinical and clinical data that should be taken into consideration by gene therapy product manufacturers in planning long-term follow-up observational studies to assess long-term adverse events.
In September 2018, the public comment period on the draft guidance was extended until December 10, 2018. Many gene therapies involve the use of viral vectors for delivery, and the future applicability of the guidance to new gene therapies that do not involve viral vectors is among the concerns raised by stakeholders. The possibility that the safety risks from long-term persistence of gene therapy products may have been overstated in the draft guidance also was raised by industry representatives. Health system pharmacists should watch for news about finalization of this FDA guidance because it may be relevant for planning for future clinical trials and commercial product implementation.
Approval by FDA currently is pending for some investigational gene therapy products, and the pace of submission of investigational new drug and biologics license applications for these products has been rapid. According to FDA Commissioner Scott Gottlieb, the agency seeks to increase its investment in gene therapy product reviewers to keep up with the many applications it receives each year. An increase in the agency budget by $50 million is needed.
The National Institutes of Health (NIH) recently committed to collaborating with FDA to streamline the federal framework and review process for gene therapy products. A focus on scientific, safety, and ethical issues will be used in attempts to reduce duplication in federal oversight. For more information, see the question and feedback below.
Published guidelines on gene therapy product development are available from the European Medicines Agency.
More Information
- U.S. Food and Drug Administration. Long term follow-up after administration of human gene therapy products—draft guidance for industry. July 6, 2018. (accessed 2019 Feb 1).
- U.S. Food and Drug Administration. Statement from FDA Commissioner Scott Gottlieb, M.D. on agency’s efforts to advance development of gene therapies. July 11, 2018. (accessed 2019 Feb 1).
- U.S. Food and Drug Administration. Statement by FDA Commissioner Scott Gottlieb, M.D., on FDA’s new steps to modernize drug development, improve efficiency and promote innovation of targeted therapies. October 15, 2018. (accessed 2019 Feb 1).
- Collins FS, Gottlieb S. The next phase of human gene-therapy oversight. N Engl J Med. 2018; 379:1393-5.
- European Medicines Agency. Guidelines relevant for advanced therapy medicinal products. (accessed 2019 Feb 1).
Feedback
According to senior leaders at FDA and NIH, all of these were factors stimulating the 2018 announcement of increased FDA-NIH collaboration in the oversight of gene therapy trials. Compared with the 1990s when FDA oversaw the first U.S. human gene therapy trial and there were many questions about the safety and efficacy of gene therapy, there is no longer sufficient evidence to claim that unique and unpredictable risks of gene therapy require oversight that falls outside the existing framework for ensuring safety. Further, the general framework for medical product safety has advanced such that tools used to address other areas of science are now well suited to gene therapy.
Over the years, much duplication in the oversight efforts of NIH, FDA, and research entities had developed, and the original need for the overlap (regulatory oversight of FDA while maintaining sponsor confidentiality, NIH’s need to conduct transparent research) was eliminated with implementation of clinical trials.gov that provides a high level of transparency. In addition, the field of gene therapy is rapidly evolving (FDA reported having more than 700 active investigational new drug applications for gene therapies in 2017), and FDA and NIH leaders recognized that changes were necessary to expedite progress safely.
Gene Therapy Resources from ASHP
To learn more about this topic, check out other free educational opportunities in this educational initiative.
- Engaging the Experts faculty interview hosted by William A. Zellmer – available now
- Live webinar, “Ask the Experts: Key Considerations in Using Viral Vector Gene Therapies” – February 21, 2019, 1.0 hr CPE, on-demand activity available May 1, 2019
- On-demand activity, “Gene Replacement and Gene Modifying Therapies: Therapeutics and Safety for Pharmacists – 1.5 hr CPE, available March 1, 2019
- Second e-newsletter – coming Spring 2019
In addition, a section of the ASHP Resource Center on Emerging Sciences addresses gene therapy and provides lists of articles, books, webinars, presentations, guidelines, policies, and other resources on the topic (ASHP membership required).
A revised policy on gene therapy developed by the ASHP Council on Pharmacy Management was approved on June 5, 2018, by the ASHP House of Delegates. The new policy advocates documentation of gene therapy in the permanent patient health record and accommodates documentation by all members of the healthcare team, including pharmacists.